fisher¶
Perform fisher’s exact test on the number of overlaps/unique intervals between 2 files.
Traditionally, in order to test whether 2 sets of intervals are related spatially, we resort to shuffling the genome and checking the simulated (shuffled) versus the observed. We can do the same analytically for many scenarios using Fisher’s Exact Test .
This implementation can calculate the number of overlaps and the number of intervals unique to each file and it infers (or accepts) the number that are not present in each file.
Given a pair of input files -a and -b in the usual BedTools parlance:
$ cat a.bed
chr1 10 20
chr1 30 40
chr1 51 52
$ cat b.bed
chr1 15 25
chr1 51 52
And a genome of 500 bases:
$ echo -e "chr1\t500" > t.genome
We may wish to know if the amount of overlap between the 2 sets of intervals is
more than we would expect given their coverage and the size of the genome. We
can do this with fisher as:
$ bedtools fisher -a a.bed -b b.bed -g t.genome
# Number of query intervals: 3
# Number of db intervals: 2
# Number of overlaps: 2
# Number of possible intervals (estimated): 37
# phyper(2 - 1, 3, 37 - 3, 2, lower.tail=F)
# Contingency Table Of Counts
#_________________________________________
# | in -b | not in -b |
# in -a | 2 | 1 |
# not in -a | 0 | 34 |
#_________________________________________
# p-values for fisher's exact test
left right two-tail ratio
1 0.0045045 0.0045045 inf
Where we can see the constructed contingency table and the pvalues for left, right and two-tail tests. From here, we can say that given 500 bases of genome, it is unlikely that we would see as many overlaps as we do if the intervals from a and b were not related.
Note
the total number of possible intervals in the above example was estimated to be 37. This is based on a heuristic that uses the mean sizes of intervals in the a and b sets and the size of the genome. The reported p-value will depend greatly on this. Below, we show how well the reported value matches with simulations.
The above had a fairly low p-value (0.0045), but if our genome were only 60 bases:
$ echo -e "chr1\t60" > t.genome
$ bedtools fisher -a a.bed -b b.bed -g t.genome
# Number of query intervals: 3
# Number of db intervals: 2
# Number of overlaps: 2
# Number of possible intervals (estimated): 4
# phyper(2 - 1, 3, 4 - 3, 2, lower.tail=F)
# Contingency Table Of Counts
#_________________________________________
# | in -b | not in -b |
# in -a | 2 | 1 |
# not in -a | 0 | 1 |
#_________________________________________
# p-values for fisher's exact test
left right two-tail ratio
1 0.5 1 inf
We can see that neither tail is significant. Intuitively, this makes sense; if we randomly place 3 intervals (from -a), and 2 (from -b) intervals from within 60 bases, it doesn’t seem unlikely that we’d see 2 overlaps.
Note also that since the genome size is much smaller, the number of possible intervals is also decreased.
Evaluation¶
The p-value depends on knowing or inferring the total number of possible intervals (to fill in the lower right corner of the contingency table). This inference is not straightforward since we will most likely have variable sized intervals within and between files. Below, we show the correspondence of the p-value reported by fisher and one from simulated data.
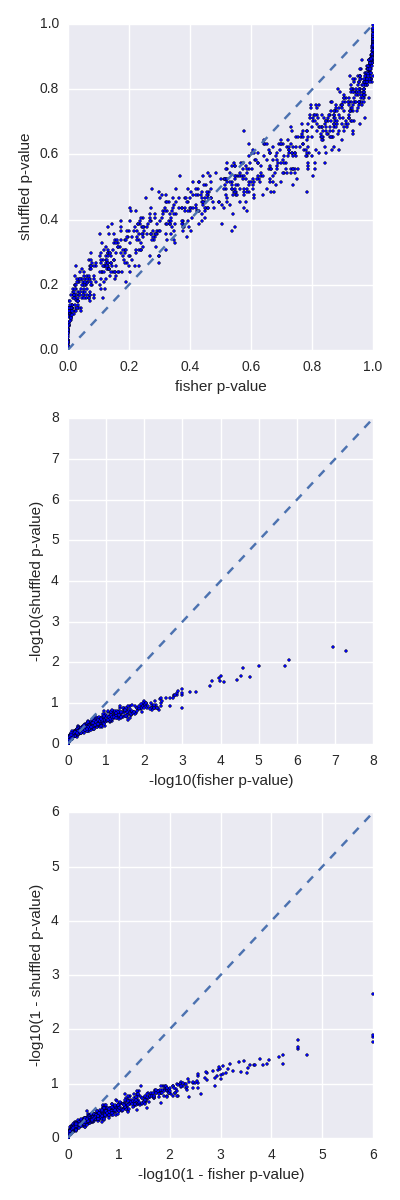
The comparison of the p-value from ‘fisher’ to one derived by simulation (see tests/fisher/ for details). The top plot shows the p-value distribution. Since we are most interested in extreme p-values, the middle plot shows -log10(p-value). The bottom plot is the same as the middle except looking at the other tail of the p-value distribution.
Note that we do see inflation from the fisher test, but we do not see ‘false-negatives’–that is, bedtools fisher is less likely to miss ‘true’ candidates, but it will give many candidates for further exploration. As such we recommend validating low p-values from fisher using simulation.
This evaluation used all known canonical genes on chromosome 1 and repeatedly (1000 times) randomly generated 3000 intervals between 20 and 5250 bases. It then calculated the p-value for each set using fisher and then using shuffled data.
Note
The fisher tool requires that your data is pre-sorted by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files).
This uses Heng Li’s implementation of Fisher’s exact test in kfunc.c.
Usage and option summary¶
Usage:
bedtools fisher [OPTIONS] -a <BED/GFF/VCF> -b <BED/GFF/VCF> -g <genome>
| Option | Description |
|---|---|
| -a | BED/GFF/VCF file A. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b | BED/GFF/VCF file B. Use “stdin” if passing B with a UNIX pipe. |
| -g | genome file listing chromosome size. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. |
