intersect¶
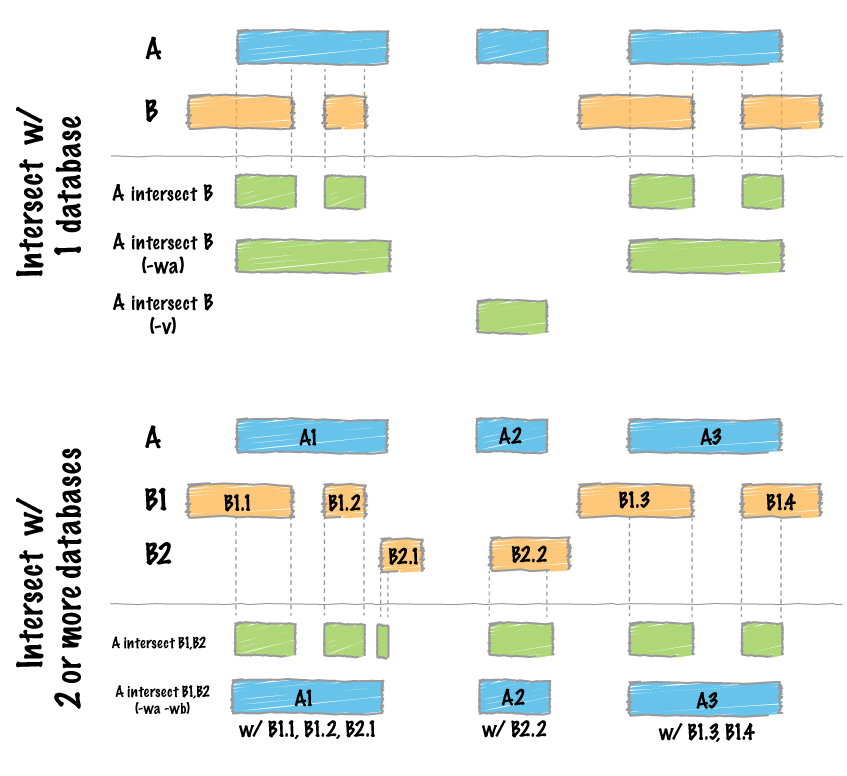
By far, the most common question asked of two sets of genomic features
is whether or not any of the features in the two sets “overlap”
with one another. This is known as feature intersection.
bedtools intersect allows one to screen for overlaps between
two sets of genomic features. Moreover, it allows one to have fine control
as to how the intersections are reported. bedtools intersect works
with both BED/GFF/VCF and BAM files as input.
Note
If you are trying to intersect very large files and are having trouble
with excessive memory usage, please presort your data by chromosome and
then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed
for BED files) and then use the -sorted option. This invokes a
memory-efficient algorithm designed for large files. This algorithm has
been substantially improved in recent (>=2.18.0) releases.
Important
As of version 2.21.0, the intersect tool can accept multiple files for the -b option. This allows one to identify overlaps between a single query (-a) file and multiple database files (-b) at once!
Usage and option summary¶
Usage:
bedtools intersect [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
(or):
intersectBed [OPTIONS] -a <FILE> \
-b <FILE1, FILE2, ..., FILEN>
| Option | Description |
|---|---|
| -a | BAM/BED/GFF/VCF file “A”. Each feature in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe. |
| -b |
NEW!!!: -b may be followed with multiple databases and/or wildcard (*) character(s). |
| -abam | BAM file A. Each BAM alignment in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe: For example: samtools view -b <BAM> | bedtools intersect -abam stdin -b genes.bed. Note: no longer necessary after version 2.19.0 |
| -ubam | Write uncompressed BAM output. The default is write compressed BAM output. |
| -bed | When using BAM input (-abam), write output as BED. The default is to write output in BAM when using -abam. For example: bedtools intersect -abam reads.bam -b genes.bed -bed |
| -wa | Write the original entry in A for each overlap. |
| -wb | Write the original entry in B for each overlap. Useful for knowing what A overlaps. Restricted by -f and -r. |
| -loj | Perform a “left outer join”. That is, for each feature in A report each overlap with B. If no overlaps are found, report a NULL feature for B. |
| -wo | Write the original A and B entries plus the number of base pairs of overlap between the two features. Only A features with overlap are reported. Restricted by -f and -r. |
| -wao | Write the original A and B entries plus the number of base pairs of overlap between the two features. However, A features w/o overlap are also reported with a NULL B feature and overlap = 0. Restricted by -f and -r. |
| -u | Write original A entry once if any overlaps found in B. In other words, just report the fact at least one overlap was found in B. Restricted by -f and -r. |
| -c | For each entry in A, report the number of hits in B while restricting to -f. Reports 0 for A entries that have no overlap with B. Restricted -f, -F, -r, and -s. |
| -C | For each entry in A, separately report the number of overlaps with each B file on a distinct line. Reports 0 for A entries that have no overlap with B. Overlaps restricted by -f, -F, -r, and -s. |
| -v | Only report those entries in A that have no overlap in B. Restricted by -f and -r. |
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. |
| -sorted | For very large B files, invoke a “sweeping” algorithm that requires position-sorted (e.g., sort -k1,1 -k2,2n for BED files) input. When using -sorted, memory usage remains low even for very large files. |
| -g | Specify a genome file the defines the expected chromosome order in the input files for use with the -sorted option. |
| -header | Print the header from the A file prior to results. |
| -names | When using multiple databases (-b), provide an alias for each that will appear instead of a fileId when also printing the DB record. |
| -filenames | When using multiple databases (-b), show each complete filename instead of a fileId when also printing the DB record. |
| -sortout | When using multiple databases (-b), sort the output DB hits for each record. |
| -nobuf | Disable buffered output. Using this option will cause each line of output to be printed as it is generated, rather than saved in a buffer. This will make printing large output files noticeably slower, but can be useful in conjunction with other software tools and scripts that need to process one line of bedtools output at a time. |
| -iobuf | Follow with desired integer size of read buffer. Optional suffixes K/M/G supported. Note: currently has no effect with compressed files. |
Default behavior¶
By default, if an overlap is found, bedtools intersect reports the shared interval between the two
overlapping features.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
Intersecting against MULTIPLE -b files.¶
As of version 2.21.0, the intersect tool can detect overlaps between a single -a file and multiple -b files (instead of just one previously). One simply provides multiple -b files on the command line.
For example, consider the following query (-a) file and three distinct (-b) files:
$ cat query.bed
chr1 1 20
chr1 40 45
chr1 70 90
chr1 105 120
chr2 1 20
chr2 40 45
chr2 70 90
chr2 105 120
chr3 1 20
chr3 40 45
chr3 70 90
chr3 105 120
chr3 150 200
chr4 10 20
$ cat d1.bed
chr1 5 25
chr1 65 75
chr1 95 100
chr2 5 25
chr2 65 75
chr2 95 100
chr3 5 25
chr3 65 75
chr3 95 100
$ cat d2.bed
chr1 40 50
chr1 110 125
chr2 40 50
chr2 110 125
chr3 40 50
chr3 110 125
$ cat d3.bed
chr1 85 115
chr2 85 115
chr3 85 115
We can now compare query.bed to all three database files at once.:
$ bedtools intersect -a query.bed \
-b d1.bed d2.bed d3.bed
chr1 5 20
chr1 40 45
chr1 70 75
chr1 85 90
chr1 110 120
chr1 105 115
chr2 5 20
chr2 40 45
chr2 70 75
chr2 85 90
chr2 110 120
chr2 105 115
chr3 5 20
chr3 40 45
chr3 70 75
chr3 85 90
chr3 110 120
chr3 105 115
Clearly this is not completely informative because we cannot tell from which file each intersection came. However, if we use -wa and -wb, this becomes abundantly clear. When these options are used, the first column after the complete -a record lists the file number from which the overlap came. The number corresponds to the order in which the files were given on the command line.
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted
chr1 1 20 1 chr1 5 25
chr1 40 45 2 chr1 40 50
chr1 70 90 1 chr1 65 75
chr1 70 90 3 chr1 85 115
chr1 105 120 2 chr1 110 125
chr1 105 120 3 chr1 85 115
chr2 1 20 1 chr2 5 25
chr2 40 45 2 chr2 40 50
chr2 70 90 1 chr2 65 75
chr2 70 90 3 chr2 85 115
chr2 105 120 2 chr2 110 125
chr2 105 120 3 chr2 85 115
chr3 1 20 1 chr3 5 25
chr3 40 45 2 chr3 40 50
chr3 70 90 1 chr3 65 75
chr3 70 90 3 chr3 85 115
chr3 105 120 2 chr3 110 125
chr3 105 120 3 chr3 85 115
In many cases, it may be more useful to report an informative “label” for each file instead of a file number. One can do this with the -names option.
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-names d1 d2 d3 \
-sorted
chr1 1 20 d1 chr1 5 25
chr1 40 45 d2 chr1 40 50
chr1 70 90 d1 chr1 65 75
chr1 70 90 d3 chr1 85 115
chr1 105 120 d2 chr1 110 125
chr1 105 120 d3 chr1 85 115
chr2 1 20 d1 chr2 5 25
chr2 40 45 d2 chr2 40 50
chr2 70 90 d1 chr2 65 75
chr2 70 90 d3 chr2 85 115
chr2 105 120 d2 chr2 110 125
chr2 105 120 d3 chr2 85 115
chr3 1 20 d1 chr3 5 25
chr3 40 45 d2 chr3 40 50
chr3 70 90 d1 chr3 65 75
chr3 70 90 d3 chr3 85 115
chr3 105 120 d2 chr3 110 125
chr3 105 120 d3 chr3 85 115
Or perhaps it may be more useful to report the file name. One can do this with the -filenames option.
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-filenames
chr1 1 20 d1.bed chr1 5 25
chr1 40 45 d2.bed chr1 40 50
chr1 70 90 d1.bed chr1 65 75
chr1 70 90 d3.bed chr1 85 115
chr1 105 120 d2.bed chr1 110 125
chr1 105 120 d3.bed chr1 85 115
chr2 1 20 d1.bed chr2 5 25
chr2 40 45 d2.bed chr2 40 50
chr2 70 90 d1.bed chr2 65 75
chr2 70 90 d3.bed chr2 85 115
chr2 105 120 d2.bed chr2 110 125
chr2 105 120 d3.bed chr2 85 115
chr3 1 20 d1.bed chr3 5 25
chr3 40 45 d2.bed chr3 40 50
chr3 70 90 d1.bed chr3 65 75
chr3 70 90 d3.bed chr3 85 115
chr3 105 120 d2.bed chr3 110 125
chr3 105 120 d3.bed chr3 85 115
Other options to intersect can be used as well. For example, let’s use -v to report those intervals in query.bed that do not overlap any of the intervals in the three database files:
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-v
chr3 150 200
chr4 10 20
Or, let’s report only those intersections where 100% of the query record is overlapped by a database record:
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-names d1 d2 d3
-f 1.0
chr1 40 45 d2 chr1 40 50
chr2 40 45 d2 chr2 40 50
chr3 40 45 d2 chr3 40 50
-wa Reporting the original A feature¶
Instead, one can force bedtools intersect to report the original “A” feature when an overlap is found. As
shown below, the entire “A” feature is reported, not just the portion that overlaps with the “B” feature.
For example:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -wa
chr1 10 20
-wb Reporting the original B feature¶
Similarly, one can force bedtools intersect to report the original “B” feature when an overlap is found. If
just -wb is used, the overlapping portion of A will be reported followed by the original “B”. If both -wa
and -wb are used, the originals of both “A” and “B” will be reported.
For example (-wb alone):
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -wb
chr1 15 20 chr1 15 20
Now -wa and -wb:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -wa -wb
chr1 10 20 chr1 15 20
-loj Left outer join. Report features in A with and without overlaps¶
By default, bedtools intersect will only report features in A that
have an overlap in B. The -loj option will report every A feature
no matter what. When there is an overlap (or more than 1), it will report
A with its overlaps. Yet when there are no overlaps, an A feature will be
reported with a NULL B feature to indicate that there were no overlaps
For example (without -loj):
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed
chr1 10 20 chr1 15 20
Now with -loj:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -loj
chr1 10 20 chr1 15 20
chr1 30 40 . -1 -1
-wo Write the amount of overlap between intersecting features¶
The -wo option reports a column after each combination of intersecting
“A” and “B” features indicating the amount of overlap in bases pairs that
is observed between the two features.
Note
When an interval in A does not intersect an interval in B, it will not be
reported. If you would like to report such intervals with an overlap equal
to 0, see the -wao option.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wo
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
-wao Write amounts of overlap for all features.¶
The -wao option extends upon the -wo option in that, unlike -wo,
it reports an overlap of 0 for features in A that do not have an intersection
in B.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wao
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
chr1 30 40 . -1 -1 0
-u (unique) Reporting the mere presence of any overlapping features¶
Often you’d like to simply know a feature in “A” overlaps one or more
features in B without reporting each and every intersection. The -u
option will do exactly this: if an one or more overlaps exists, the
A feature is reported. Otherwise, nothing is reported.
For example, without -u:
$ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
chr1 17 20
Now with -u:
$ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed -u
chr1 10 20
-c Reporting the number of overlapping features¶
The -c option reports a column after each “A” feature indicating the number (0 or more) of overlapping features found in “B”. Therefore, each feature in A is reported once.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -c
chr1 10 20 2
chr1 30 40 0
The -c option can also work with multiple -B files. In this case, the reported count will reflect the total number of intersections observed across _all_ -B files. It will not report a separate count for each database file. This can be achieved with the -C option.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ cat C.bed
chr1 16 21
chr1 19 26
$ bedtools intersect -a A.bed -b B.bed -c
chr1 10 20 4
chr1 30 40 0
-C Reporting the number of overlapping features for each database file¶
Unlike the the -c option, in the case of multiple -B files, the -C option will report a separate count for each database file.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ cat C.bed
chr1 16 21
chr1 19 26
$ bedtools intersect -a A.bed -b B.bed -C
chr1 10 20 1 2
chr1 10 20 2 2
chr1 30 40 1 0
chr1 30 40 1 0
If you would like to see more useful information than the file _number_ from which the counts came, one can use the -filenames or -names options.
$ bedtools intersect -a A.bed -b B.bed -C -filenames
chr1 10 20 A.bed 2
chr1 10 20 B.bed 2
chr1 30 40 A.bed 0
chr1 30 40 B.bed 0
$ bedtools intersect -a A.bed -b B.bed -C -names a b
chr1 10 20 a 2
chr1 10 20 b 2
chr1 30 40 a 0
chr1 30 40 b 0
-v Reporting the absence of any overlapping features¶
There will likely be cases where you’d like to know which “A” features
do not overlap with any of the “B” features. Perhaps you’d like to know
which SNPs don’t overlap with any gene annotations. The -v
(an homage to “grep -v”) option will only report those “A” features
that have no overlaps in “B”.
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed -v
chr1 30 40
-f Requiring a minimal overlap fraction¶
By default, bedtools intersect will report an overlap between A and B so long as there is at least one base
pair is overlapping. Yet sometimes you may want to restrict reported overlaps between A and B to cases
where the feature in B overlaps at least X% (e.g. 50%) of the A feature. The -f option does exactly
this.
For example (note that the second B entry is not reported):
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 130 201
chr1 180 220
$ bedtools intersect -a A.bed -b B.bed -f 0.50 -wa -wb
chr1 100 200 chr1 130 201
-r, and -f Requiring reciprocal minimal overlap fraction¶
Similarly, you may want to require that a minimal fraction of both the A and the B features is overlapped. For example, if feature A is 1kb and feature B is 1Mb, you might not want to report the overlap as feature A can overlap at most 1% of feature B. If one set -f to say, 0.02, and one also enable the -r (reciprocal overlap fraction required), this overlap would not be reported.
For example (note that the second B entry is not reported):
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 130 201
chr1 130 200000
$ bedtools intersect -a A.bed -b B.bed -f 0.50 -r -wa -wb
chr1 100 200 chr1 130 201
-s Enforcing same strandedness¶
By default, bedtools intersect will report overlaps between features
even if the features are on opposite strands. However, if strand information
is present in both BED files and the “-s” option is used, overlaps will only
be reported when features are on the same strand.
For example (note that the first B entry is not reported):
$ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -s
chr1 100 200 a1 100 + chr1 132 203 b2 100 +
-S Enforcing opposite “strandedness”¶
The -s option enforces that overlaps be on the same strand. In some
cases, you may want to enforce that overlaps be found on opposite strands.
In this, case use the -S option.
For example:
$ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -S
chr1 100 200 a1 100 + chr1 130 201 b1 100 -
-abam Default behavior when using BAM input (deprecated since 2.18.0)¶
When comparing alignments in BAM format (-abam) to features in BED format (-b), bedtools intersect
will, by default, write the output in BAM format. That is, each alignment in the BAM file that meets
the user’s criteria will be written (to standard output) in BAM format. This serves as a mechanism to
create subsets of BAM alignments are of biological interest, etc. Note that only the mate in the BAM
alignment is compared to the BED file. Thus, if only one end of a paired-end sequence overlaps with a
feature in B, then that end will be written to the BAM output. By contrast, the other mate for the
pair will not be written. One should use pairToBed(Section 5.2) if one wants each BAM alignment
for a pair to be written to BAM output.
$ bedtools intersect -abam reads.unsorted.bam -b simreps.bed | \
samtools view - | \
head -3
BERTHA_0001:3:1:15:1362#0 99 chr4 9236904 0 50M = 9242033 5 1 7 9
AGACGTTAACTTTACACACCTCTGCCAAGGTCCTCATCCTTGTATTGAAG W c T U ] b \ g c e g X g f c b f c c b d d g g V Y P W W _
\c`dcdabdfW^a^gggfgd XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:19 X1:i:2 XM:i:0 XO:i:0 XG:i:0 MD:Z:50
BERTHA _0001:3:1:16:994#0 83 chr6 114221672 37 25S6M1I11M7S =
114216196 -5493 G A A A G G C C A G A G T A T A G A A T A A A C A C A A C A A T G T C C A A G G T A C A C T G T T A
gffeaaddddggggggedgcgeggdegggggffcgggggggegdfggfgf XT:A:M NM:i:3 SM:i:37 AM:i:37 XM:i:2 X O : i :
1 XG:i:1 MD:Z:6A6T3
BERTHA _0001:3:1:16:594#0 147 chr8 43835330 0 50M =
43830893 -4487 CTTTGGGAGGGCTTTGTAGCCTATCTGGAAAAAGGAAATATCTTCCCATG U
\e^bgeTdg_Kgcg`ggeggg_gggggggggddgdggVg\gWdfgfgff XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:10 X1:i:7 X M : i :
2 XO:i:0 XG:i:0 MD:Z:1A2T45
Note
As of version 2.18.0, it is no longer necessary to specify a BAM input file via -abam.
Bedtools now autodetects this when -a is used.
-ubam Default behavior when using BAM input¶
The -ubam option writes uncompressed BAM output to stdout. This is
useful for increasing the speed of pipelines that accept the output of
bedtools intersect as input, since the receiving tool does not need to
uncompress the data.
-bed Output BED format when using BAM input¶
When comparing alignments in BAM format (-abam) to features in BED format (-b), bedtools intersect
will optionally write the output in BED format. That is, each alignment in the BAM file is converted
to a 6 column BED feature and if overlaps are found (or not) based on the user’s criteria, the BAM
alignment will be reported in BED format. The BED “name” field is comprised of the RNAME field in
the BAM alignment. If mate information is available, the mate (e.g., “/1” or “/2”) field will be
appended to the name. The “score” field is the mapping quality score from the BAM alignment.
$ bedtools intersect -abam reads.unsorted.bam -b simreps.bed -bed | head -20
chr4 9236903 9236953 BERTHA_0001:3:1:15:1362#0/1 0 +
chr6 114221671 114221721 BERTHA_0001:3:1:16:994#0/1 37 -
chr8 43835329 43835379 BERTHA_0001:3:1:16:594#0/2 0 -
chr4 49110668 49110718 BERTHA_0001:3:1:31:487#0/1 23 +
chr19 27732052 27732102 BERTHA_0001:3:1:32:890#0/2 46 +
chr19 27732012 27732062 BERTHA_0001:3:1:45:1135#0/1 37 +
chr10 117494252 117494302 BERTHA_0001:3:1:68:627#0/1 37 -
chr19 27731966 27732016 BERTHA_0001:3:1:83:931#0/2 9 +
chr8 48660075 48660125 BERTHA_0001:3:1:86:608#0/2 37 -
chr9 34986400 34986450 BERTHA_0001:3:1:113:183#0/2 37 -
chr10 42372771 42372821 BERTHA_0001:3:1:128:1932#0/1 3 -
chr19 27731954 27732004 BERTHA_0001:3:1:130:1402#0/2 0 +
chr10 42357337 42357387 BERTHA_0001:3:1:137:868#0/2 9 +
chr1 159720631 159720681 BERTHA_0001:3:1:147:380#0/2 37 -
chrX 58230155 58230205 BERTHA_0001:3:1:151:656#0/2 37 -
chr5 142612746 142612796 BERTHA_0001:3:1:152:1893#0/1 37 -
chr9 71795659 71795709 BERTHA_0001:3:1:177:387#0/1 37 +
chr1 106240854 106240904 BERTHA_0001:3:1:194:928#0/1 37 -
chr4 74128456 74128506 BERTHA_0001:3:1:221:724#0/1 37 -
chr8 42606164 42606214 BERTHA_0001:3:1:244:962#0/1 37 +
-split Reporting overlaps with spliced alignments or blocked BED features¶
As described in section 1.3.19, bedtools intersect will, by default, screen for overlaps against the entire span of a spliced/split BAM alignment or blocked BED12 feature. When dealing with RNA-seq reads, for example, one typically wants to only screen for overlaps for the portions of the reads that come from exons (and ignore the interstitial intron sequence). The -split command allows for such overlaps to be performed.
For example, the diagram below illustrates the default behavior. The blue dots represent the “split/ spliced” portion of the alignment (i.e., CIGAR “N” operation). In this case, the two exon annotations are reported as overlapping with the “split” BAM alignment, but in addition, a third feature that overlaps the “split” portion of the alignment is also reported.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Exons --------------- ----------
BED/BAM A ************.......................................****
BED File B ^^^^^^^^^^^^^^^ ^^^^^^^^ ^^^^^^^^^^
Result =============== ======== ==========
In contrast, when using the -split option, only the exon overlaps are reported.
Chromosome ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Exons --------------- ----------
BED/BAM A ************.......................................****
BED File B ^^^^^^^^^^^^^^^ ^^^^^^^^ ^^^^^^^^^^
Result =============== ==========
-sorted Invoke a memory-efficient algorithm for very large files.¶
The default algorithm for detecting overlaps loads the B file into an R-tree
structure in memory. While fast, it can consume substantial memory for large
files. For these reason, we provide an alternative, memory efficient algorithm
that depends upon inout files that have been sorted by chromosome and then by
start position. When both input files are position-sorted, the algorithm can
“sweep” through the data and detect overlaps on the fly in a manner much
like the way database systems join two tables. This option is invoked with the
-sorted option.
Note
By default, the -sorted option requires that the records are GROUPED
by chromosome and that within each chromosome group, the records are sorted by
chromosome position. One way to achieve this (for BED files for example) is use
the UNIX sort utility to sort both files by chromosome and then by position.
That is, sort -k1,1 -k2,2n in.bed > in.sorted.bed. However, since we merely
require that the chromsomes are grouped (that is, all records for a given chromosome
come in a single block in the file), sorting criteria other than the alphanumeric
criteria that is used by the sort utility are fine. For example, you could use
the “version sort” (-V) option in newer versions of GNU sort to make the chromosomes
come in this (chr1, chr2, chr3) order instead of this (chr1, chr10, chr11) order.
For example:
$ bedtools intersect -a big.sorted.bed -b huge.sorted.bed -sorted
-g Define an alternate chromosome sort order via a genome file.¶
As described above, the -sorted option expects that the input files are grouped
by chromosome. However, there arise cases where ones input
files are sorted by a different criteria and it is to computationally onerous
to resort the files alphanumerically. For example, the GATK expects that
BAM files are sorted in a very specific manner. The -g option allows
one to specify an exact ording that should be expected in the input (e.g.,
BAM, BED, etc.) files. All you need to do is re-order you genome file to
specify the order. Also, the use of a genome file to specify the expected
order allows the intersect tool to detect when two files are internally
grouped but each file actually follows a different order. This will cause
incorrect results and the -g file will alert you to such problems.
For example, an alphanumerically ordered genome file would look like the following:
$ cat hg19.genome
chr1 249250621
chr10 135534747
chr11 135006516
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
chr18 78077248
chr19 59128983
chr2 243199373
chr20 63025520
chr21 48129895
chr22 51304566
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chr8 146364022
chr9 141213431
chrM 16571
chrX 155270560
chrY 59373566
However, if your input BAM or BED files are ordered such as chr1, chr2, chr3, etc.,
one need to simply reorder the genome file accordingly:
$ sort -k1,1V hg19.genome > hg19.versionsorted.genome
$ cat hg19.versionsorted.genome
chr1 249250621
chr2 243199373
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chr8 146364022
chr9 141213431
chr10 135534747
chr11 135006516
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
chr18 78077248
chr19 59128983
chr20 63025520
chr21 48129895
chr22 51304566
chrM 16571
chrX 155270560
chrY 59373566
At this point, one can now use the -sorted option along with the genome file
in order to properly process the input files that abide by something other than an
alphanumeric sorting order.
$ bedtools intersect -a a.versionsorted.bam -b b.versionsorted.bed \
-sorted \
-g hg19.versionsorted.genome
Et voila.
-header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.
