summary¶
Genomics experiments have numerous sources of both technical and biological variation
that can confound analysis and interpretation. Therefore, one of the most important steps
in genomics data analysis is generating high-level summary stats of one’s data to ask if the
basic observations are in line with the expectation. Doing such quality control as early
as possible in the analysis workflow helps to head off unnecessary time spent chasing
technical artifacts that masquerade as biological signal. This quality control
is the motivation behind the bedtools summary command.
Given an input interval file in standard formats, as well as a genome file defining
the chromosome names and lengths relevant to your data, bedtools summary will compute
a number of summary statistics detailing, for each chromosome, the number of intervals,
the total number of base pairs, and the fraction of intervals and base pairs observed
in your input file. From these summary measures, one can get a quick sense of questions like:
- Are all chromosomes represented in my data or annotation file?
- Do all chromosomes have the expected number of intervals or fraction of base pairs represented?
- Which chromosomes are outliers?
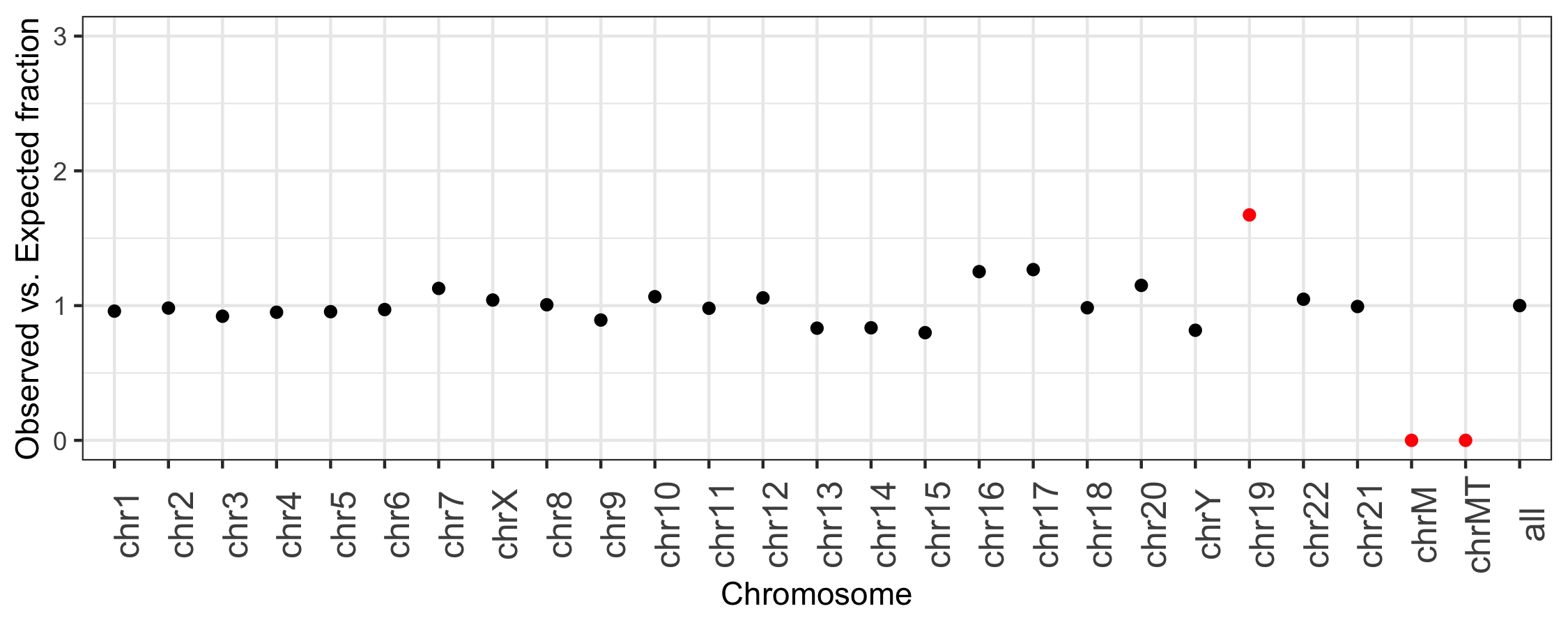
For example, the following plot was generated directly from the output of bedtools summary.
It depicts, for each chromosome, the the fraction of all intervals in the RepeatMasker track from UCSC
observed on each chromosome versus the fraction of RepeatMasker intervals that are expected
for each chromosome, based on the fraction of the genome that each chromosome represents.
This plot highlights that chr19, chrM, and chrMT (the different mitochindrial reference genomes) are outliers.
Chromosome 19 has more than 1.5 times the intervals that are expected based upon the length of
the chromosome. ChrM has no intervals, making the observed to expected ratio be 0.
This former is because the repeat content of chromosome 19 “is approximately 55%, more than 10% higher
than the genome-wide average” (Grimwood J, et al. Nature. 2004;428:529–35). The latter because
are indeed no repeat annotations provided by UCSC for the mitochondrial genome.
In this case, the extremes in obersved versus expected ratios make sense. However, this tool provides the ability to detect cases that do not and are either artifacts or biological signal.
Default behavior¶
bedtools summary scans the input interval file and the chromosome lengths provided in
the “genome” file and reports the following columns for each chromosome.
- chrom (chromosome name)
- chrom_length (the length of the chromosome in bp)
- num_ivls (the total number of intervals observed for the chromosome)
- total_ivl_bp (the total number of bp observed in the intervals for the chromosome)
- chrom_frac_genome (the fraction of the genome represented by the chromosome)
- frac_all_ivls (the fraction of all intervals observed the chromosome)
- frac_all_bp (the fraction of all bp observed the chromosome)
- min (the smallest interval observed on that chromosome)
- max (the largest interval observed on that chromosome)
- mean (the mean interval length observed on that chromosome)
In addition, bedtools summary reports a final line with the chromosome name “all”, which
depicts the metrics above but tabulated across all chromosomes in the input file.
As an example, let’s recreate the figure above by running bedtools summary on the
simple repeats track from UCSC. We’ll begin by downloading the data in track the
track table and converting it to BED format.
curl -s http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/simpleRepeat.txt.gz \
| gzcat \
| cut -f 2-5 \
| grep -v -E 'Un|fix|random|alt|hap' \
> simrep.grch38.bed
head simrep.grch38.bed
chr1 10000 10468 trf
chr1 10627 10800 trf
chr1 10757 10997 trf
chr1 11225 11447 trf
chr1 11271 11448 trf
chr1 11283 11448 trf
chr1 19305 19443 trf
chr1 20828 20863 trf
chr1 30862 30959 trf
chr1 44835 44876 trf
Now, let’s make a “genome” file for GRCh38 from the chromInfo table at UCSC
curl -s http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/chromInfo.txt.gz \
| gzcat \
| cut -f 1-2 \
| grep -v -E 'Un|fix|random|alt|hap' \
> grch38.genome.txt
head grch38.genome.txt
chr1 249250621
chr2 243199373
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chrX 155270560
chr8 146364022
chr9 141213431
Now, let’s run bedtools summary.
bedtools summary -i simrep.grch38.bed -g grch38.genome.txt | column -t
chrom chrom_length num_ivls total_ivl_bp chrom_frac_genome frac_all_ivls frac_all_bp min max mean
chr1 249250621 74548 15557884 0.080514834 0.077210928 0.048725518 25 124438 208.696195740
chr2 243199373 74474 14493548 0.078560114 0.077134284 0.045392139 25 336509 194.612186803
chr3 198022430 56894 13946854 0.063966714 0.058926309 0.043679955 25 500000 245.137518895
chr4 191154276 56685 10160257 0.061748110 0.058709844 0.031820766 25 136950 179.240663315
chr5 180915260 53887 16801740 0.058440625 0.055811896 0.052621133 25 500000 311.795794904
chr6 171115067 51802 11222841 0.055274892 0.053652418 0.035148658 25 500000 216.648797344
chr7 159138663 55972 20054618 0.051406183 0.057971375 0.062808775 25 150228 358.297327235
chrX 155270560 50432 27398336 0.050156679 0.052233481 0.085808462 25 500000 543.272842640
chr8 146364022 45937 15650021 0.047279621 0.047577915 0.049014080 25 500000 340.684437382
chr9 141213431 39329 10932158 0.045615838 0.040733870 0.034238272 25 159861 277.966843805
chr10 135534747 45074 11407694 0.043781466 0.046684087 0.035727596 25 110000 253.088121755
chr11 135006516 41279 14127024 0.043610833 0.042753526 0.044244227 25 500000 342.232709126
chr12 133851895 44151 13878240 0.043237859 0.045728117 0.043465064 25 356015 314.335802134
chr13 115169878 29907 9423815 0.037203051 0.030975307 0.029514313 25 110000 315.103989033
chr14 107349540 27973 9245970 0.034676866 0.028972223 0.028957323 25 173523 330.531941515
chr15 102531392 25557 9565023 0.033120471 0.026469921 0.029956561 25 110000 374.262354736
chr16 90354753 35288 11959674 0.029187080 0.036548522 0.037456334 25 138208 338.916175470
chr17 81195210 32093 16264416 0.026228295 0.033239393 0.050938295 25 132210 506.790141152
chr18 78077248 23966 18684937 0.025221107 0.024822089 0.058519091 25 500000 779.643536677
chr20 63025520 22608 12620160 0.020358983 0.023415580 0.039524901 25 500000 558.216560510
chrY 59373566 15130 4564760 0.019179301 0.015670458 0.014296307 25 227093 301.702577660
chr19 59128983 30854 11391752 0.019100294 0.031956135 0.035677667 25 396802 369.214753355
chr22 51304566 16760 10691540 0.016572792 0.017358684 0.033484683 25 498537 637.920047733
chr21 48129895 14911 9253172 0.015547285 0.015443636 0.028979879 25 499939 620.560123399
chrM 16571 0 0 0.000005353 0.000000000 0.000000000 -1 -1 -1
chrMT 16569 0 0 0.000005352 0.000000000 0.000000000 -1 -1 -1
all 3095710552 965511 319296434 1.0 1.0 1.0 25 500000 330.702015824
Notice the following:
- There are 0 intervals reported for chrM or chrMT; therefore, the min, max, and mean are all “-1”.
- The last line in the output is has an “genome” chromosome, meaning it is a summary of all of the chromosomes.
Using this report, there are many high-level sanity checks one can explore. For example, we can create the plot described above by saving the output to a file.
bedtools summary -i simrep.grch38.bed -g grch38.genome.txt > ~/simrep.summary.tsv
Now run following R code. (Sorry, I am not an R expert)
if (!require("dplyr")) install.packages("dplyr")
if (!require("ggplot2")) install.packages("ggplot2")
library(dplyr)
library(ggplot2)
x = read_tsv('~/simrep.summary.tsv')
x = x %>% mutate(obs_v_exp = frac_all_ivls/chrom_frac_genome)
p = ggplot(x) +
ylim(0,3) +
ylab("Observed vs. Expected fraction") +
xlab("Chromosome") +
geom_point(aes(x=factor(chrom, level=chrom),
y=obs_v_exp,
color=ifelse(obs_v_exp>1.5 | obs_v_exp<0.5, 'red', 'black'))) +
scale_color_identity() +
theme_bw()
p + theme(axis.text.x = element_text(size = 12, angle = 90))
