Release History¶
Version 2.30.0 (23-Dec-2021)¶
- Thanks to Hao Hou (github: @38), we have substantial improvements in the speed associated with parsing input files and in printing results. It turns our that these tasks consume a large proportion of run time, especially as input files increase in size. These changes result in a 2-3X improvement in speed, depending on input types, options, etc.
- Thanks to John Marshall (github: @jmarshall), who improved the stability and cleanliness of the code used for random number generation. These changes also squash a bug that arises on Debian systems.
- John Marshall cleaned up some lingering data type problems in the slop tool.
- Thanks to @gringer for adding teh -ignoreD option to the genomecov tool, which allows D CIGAR operations to be ignored when calculating coverage. This is useful for long-read technologies with high INDEL error rates.
- Added a fix for a bug that did not properly handle the splitting of intervals in BED12 records with one block.
- Thanks to John Marshall (github: @jmarshall), we have addressed numerical instability issues in the fisher tool.
- Thanks to Hao Hou (github: @38), reference genomes can be read as an environment variable (CRAM_REFERENCE) when using CRAM input files.
- Added a -rna option to the getfasta tool to allow support for RNA genomes.
- Thanks to Hao Hou (github: @38), we fixed input file format detection bugs arising in ZSH.
- Thanks to Josh Shapiro (github:@jashapiro) for clarifying a confusing inconcistency in the documentation for the coverage tool.
- Thanks to Hao Hou (github: @38), we suppressed unnecessary warnings when reading GZIPP’ed files.
- Thanks to Hao Hou (github: @38), we fixed an overflow bug in the shuffle tool.
- Thanks to Hao Hou (github: @38), we fixed an data type bug in the shift tool.
- Thanks to John Marshall (github: @jmarshall) and Hao Hou (github: @38), we have cleaned up the internal support for htslib.
Version 2.29.2 (17-Dec-2019)¶
- Fixed a bug that mistakenly removed a BAM/CRAM header line (sorting criteria).
Version 2.29.1 (9-Dec-2019)¶
- Fixed a bug that now allows blocked intersection to be counted based on unique base pairs of overlap. The resolution for issue 750 in version 2.29.0 mistakenly allowed for fractional overlap to be counted based upon redundant overlap.
- Moved to Github Continuous Integration for automatic testing.
- Fixed a bug that injected erroneous quality values with BAM records had no valid quality values.
- Fixed a bug that destroyed backwards compatibility in the getfasta tool. Thanks to Torsten Seeman for reporting this.
- Fixed a corner case bug in the reldist tool.
- Fixed a bug in the bedtobam tool that caused the last character in read names to be removed.
- Fixed a bug causing a segfault in the jaccard tool.
- Fixed a bug causing a corner case issue in the way coordinates are reported in the flank tool.
Version 2.29.0 (3-Sep-2019)¶
- Added a new -C option to the intersect tool that separately reports the count of intersections observed for each database (-b) file given. Formerly, the -c option reported to sum of all intersections observed across all database files.
- Fixed an important bug in intersect that prevented some split reads from being counted properly with using the -split option with the -f option.
- Fixed a bug in shuffle such that shuffled regions should have the same strand as the chose -incl region.
- Added a new -L option to L`imit the output of the `complement tool to solely the chromosomes that are represented in the -i file.
- Fixed a regression in the multicov tool introduced in 2.28 that caused incorrect counts.
- Added support for multi-mapping reads in the bamtofastq tool.
- Fixed a bug that prevented the “window” tool from properly adding interval “slop” to BAM records.
- Fixed a bug that caused the slop tool to not truncate an interval’s end coordinate when it overlapped the end of a chromosome.
- Added support for the “=” and “X” CIGAR operations to bamtobed.
- Various other minor bug fixes and improvements to documentation.
Version 2.28.0 (23-Mar-2019)¶
- Included support for htslib to enable CRAM support and long-term stability (Thanks to Hao Hou!)
- Included support for genomes with large chromosomes by moving to 64-bit integeres throughout the code base. Thanks to Brent Pedersen and John Marshall!
- We now provide a statically-linked binary for LINUX (not OSX) systems.
- Various minor bug fixes.
Version 2.27.0 (6-Dec-2017)¶
- Fixed a big memory leak and algorithmic flaw in the split option. Thanks to Neil Kindlon!
- Resolved compilation errors on OSX High Sierra. Many thanks to @jonchang!
- Fixed a bug in the shift tool that caused some intervals to exceed the end of the chromosome. Thanks to @wlholtz
- Fixed major bug in groupby that prevented proper functionality.
- Speed improvements to the shuffle tool.
- Bug fixes to the p-value calculation in the fisher tool. Thanks to Brent Pedersen.
- Allow BED headers to start with chrom or chr
- Fixes to the “k-closest” functionality in the closest tool. Thanks to Neil Kindlon.
- Fixes to the output of the freqasc, freqdesc, distinct_sort_num and distinct_sort, and num_desc operations in the groupby tool. Thanks to @ghuls.
- Many minor bug fixes and compilation improvements from Luke Goodsell.
- Added the -fullHeader option to the maskfasta tool. Thanks to @ghuls.
- Many bug fixes and performance improvements from John Marshall.
- Fixed bug in the -N/-f behavior in subtract.
- Full support for .fai files as genome (-g) files.
- Many other minor bug fixes and functionality improvements.
Version 2.26.0 (7-July-2016)¶
- Fixed a major memory leak when using
-sorted. Thanks to Emily Tsang and Stephen Montgomery. - Fixed a bug for BED files containing a single record with no newline. Thanks to @jmarshall.
- Fixed a bug in the contigency table values for thr
fishertool. - The
getfastatool includes name, chromosome and position in fasta headers when the-nameoption is used. Thanks to @rishavray. - Fixed a bug that now forces the
coveragetool to process every record in the-afile. - Fixed a bug preventing proper processing of BED files with consecutive tabs.
- VCF files containing structural variants now infer SV length from either the SVLEN or END INFO fields. Thanks to Zev Kronenberg.
- Resolve off by one bugs when intersecting GFF or VCF files with BED files.
- The
shuffletool now uses roulette wheel sampling to shuffle to-inclregions based upon the size of the interval. Thanks to Zev Kronenberg and Michael Imbeault. - Fixed a bug in
coveragethat prevented correct calculation of depth when using the-splitoption. - The
shuffletool warns when an interval exceeds the maximum chromosome length. - The
complementtool better checks intervals against the chromosome lengths. - Fixes for
stddev,min, andmaxoperations. Thanks to @jmarshall. - Enabled
stdev,sstdev,freqasc, andfreqdescoptions forgroupby. - Allow
-sand-wto be used in any order formakewindows. - Added new
-bedOutoption togetfasta. - The
-roption forces the-Fvalue forintersect. - Add
-pcoption to thegenomecovtool, allowing coverage to be calculated based upon paired-end fragments.
Version 2.25.0 (3-Sept-2015)¶
- Added new -F option that allows one to set the minimum fraction of overlap required for the B interval. This complements the functionality of the -f option.Available for intersect, coverage, map, subtract, and jaccard.
- Added new -e option that allows one to require that the minimum fraction overlap is achieved in either A _OR_ B, not A _AND_ B which is the behavior of the -r option. Available for intersect, coverage, map, subtract, and jaccard.
- Fixed a longstanding bug that prevented genomecov from reporting chromosomes that lack a single interval.
- Modified a src directory called “aux” to “driver” to prevent compilation errors on Windows machines. Thanks very much to John Marshall.
- Fixed a regression that caused the coverage tool to complain if BED files had less than 5 columns.
- Fixed a variable overload bug that prevented compilation on Debian machines.
- Speedups to the groupby tool.
- New -delim option for the groupby tool.
- Fixed a bug in map that prevented strand-specifc overlaps from being reported when using certain BEDPLUS formats.
- Prevented excessive memory usage when not using pre-sorted input.
Version 2.24.0 (27-May-2015)¶
- The coverage tool now takes advantage of pre-sorted intervals via the -sorted option. This allows the coverage tool to be much faster, use far less memory, and report coverage for intervals in their original order in the input file.
- We have changed the behavior of the coverage tool such that it is consistent with the other tools. Specifically, coverage is now computed for the intervals in the A file based on the overlaps with the B file, rather than vice versa.
- The
subtracttool now supports pre-sorted data via the-sortedoption and is therefore much faster and scalable. - The
-nonamecheckoption provides greater tolerance for chromosome labeling when using the-sortedoption. - Support for multiple SVLEN tags in VCF format, and fixed a bug that failed to process SVLEN tags coming at the end of a VCF INFO field.
- Support for reverse complementing IUPAC codes in the
getfastatool. - Provided greater flexibility for “BED+” files, where the first 3 columns are chrom, start, and end, and the remaining columns are free-form.
- We now detect stale FAI files and recreate an index thanks to a fix from @gtamazian.
- New feature from Pierre Lindenbaum allowing the
sorttool to sort files based on the chromosome order in afaidxfile. - Eliminated multiple compilation warnings thanks to John Marshall.
- Fixed bug in handling INS variants in VCF files.
Version 2.23.0 (22-Feb-2015)¶
- Added
-koption to the closest tool to report the k-closest features in one or more -b files. - Added
-fdoption to the closest tool to for the reporting of downstream features in one or more -b files. Requires -D to dictate how “downstream” should be defined. - Added
-fuoption to the closest tool to for the reporting of downstream features in one or more -b files. Requires -D to dictate how “downstream” should be defined. - Pierre Lindenbaum added a new split tool that will split an input file into multiple sub files. Unlike UNIX split, it can balance the chunking of the sub files not just by number of lines, but also by total number of base pairs in each sub file.
- Added a new spacing tool that reports the distances between features in a file.
- Jay Hesselberth added a
-reverseoption to the makewindows tool that reverses the order of the assigned window numbers. - Fixed a bug that caused incorrect reporting of overlap for zero-length BED records. Thanks to @roryk.
- Fixed a bug that caused the map tool to not allow
-bto be specified before-a. Thanks to @semenko. - Fixed a bug in
makewindowsthat mistakenly required-swith-n.
Version 2.22.1 (01-Jan-2015)¶
- When using -sorted with intersect, map, and closest, bedtools can now detect and warn you when your input datasets employ different chromosome sorting orders.
- Fixed multiple bugs in the new, faster closest tool. Specifically, the -iu, -id, and -D options were not behaving properly with the new “sweeping” algorithm that was implemented for the 2.22.0 release. Many thanks to Sol Katzman for reporting these issues and for providing a detailed analysis and example files.
- We FINALLY wrote proper documentation for the closest tool (http://bedtools.readthedocs.org/en/latest/content/tools/closest.html)
- Fixed bug in the tag tool when using -intervals, -names, or -scores. Thanks to Yarden Katz for reporting this.
- Fixed issues with chromosome boundaries in the slop tool when using negative distances. Thanks to @acdaugherty!
- Multiple improvements to the fisher tool. Added a -m option to the fisher tool to merge overlapping intervals prior to comparing overlaps between two input files. Thanks to@brentp
- Fixed a bug in makewindows tool requiring the use of -b with -s.
- Fixed a bug in intersect that prevented -split from detecting complete overlaps with -f 1. Thanks to @tleonardi .
- Restored the default decimal precision to the groupby tool.
- Added the -prec option to the merge and map tools to specific the decimal precision of the output.
Version 2.22.0 (12-Nov-2014)¶
- The “closest” tool now requires sorted files, but this requirement now enables it to simultaneously find the closest intervals from many (not just one) files.
- We now have proper support for “imprecise” SVs in VCF format. This addresses a long standing (sorry) limitation in the way bedtools handles VCF files.
Version 2.21.0 (18-Sep-2014)¶
- Added ability to intersect against multiple -b files in the intersect tool.
- Fixed a bug causing slowdowns in the -sorted option when using -split with very large split alignments.
- Added a new fisher tool to report a P-value associated with the significance of the overlaps between two interval sets. Thanks to @brentp!
- Added a “genome” file for GRCh38. Thanks @martijnvermaat!
- Fixed a bug in the -pct option of the slop tool. Thanks to @brentp!
- Tweak to the Makefile to accomodate Intel compilers. Thanks to @jmarshall.
- Many updates to the docs from the community. Thank you!
Version 2.20.1 (23-May-2014)¶
- Fixed a float rounding bug causing occassional off-by-one issues in the slop added by the
sloptool. Thanks to @slw287r. - Fixed a bug injected in 2.19 arising when files have a single line not ending in a newline. Thanks to @cwarden45.
Version 2.20.0 (22-May-2014)¶
- The
mergetool now supports BAM input.
- The
-n,-nms, and-scoresoptions are deprecated in favor of the new, substantially more flexible,-cand-ooptions. See the docs.- It now supports the
-headeroption.- It now supports the
-Soption.
- The
maptool now supports BAM input. - The
jaccardtool is now ~3 times faster.
- It now supports the
-splitoption.- It now supports the
-soption.- It now supports the
-Soption.
- We have fixed several CLANG compiler issues/ Thanks to John Marshall for the thorough report.
- We added support for “X” and “=” CIGAR operators. Thanks to Pierre Lindenbaum.
- Fixed bugs for empty files.
- Improved the
-incloption in theshuffletool such that the distibution is much more random. - Fixed a bug in
slopwhen very large slop values are used.
Version 2.19.1 (6-Mar-2014)¶
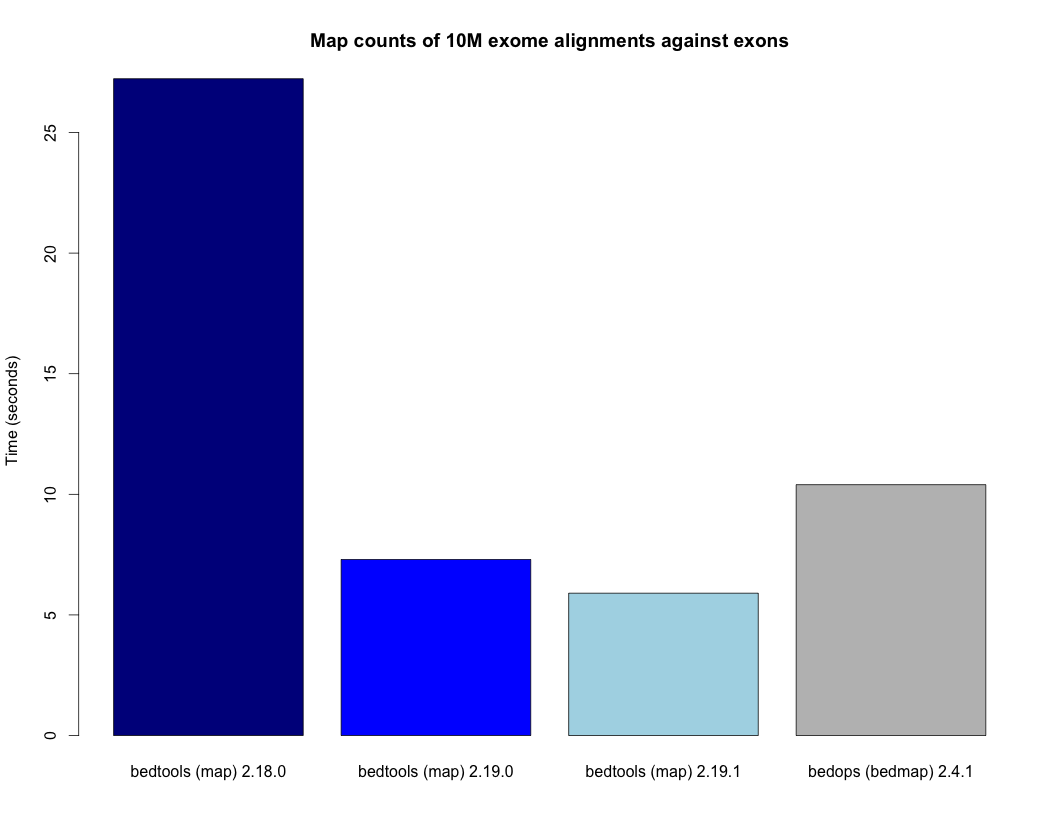
- Bug fix to intersect causing BAM footers to be erroneously written when -b is BAM
- Speedup for the map tool. - http://bedtools.readthedocs.org/en/latest/_images/map-speed-comparo.png
- Map tool now allows multiple columns and operations in a single run. - http://bedtools.readthedocs.org/en/latest/content/tools/map.html#multiple-operations-and-columns-at-the-same-time
Version 2.19.0 (8-Feb-2014)¶
Bug Fixes¶
- Fixed a long standing bug in which the number of base pairs of overlap was incorrectly calculated when using the -wo option with the -split option. Thanks to many for reporting this.
- Fixed a bug in which certain flavors of unmapped BAM alignments were incorrectly rejected in the latest 2.18.* series. Thanks very much to Gabriel Pratt.
Enhancements¶
- Substantially reduced memory usage, especially when dealing with unsorted data. Memory usage ballooned in the 2.18.* series owing to default buffer sizes we were using in a custom string class. We have adjusted this and the memory usage has returned to 2.17.* levels while maintaining speed increases. Thanks so much to Ian Sudberry rightfully complaining about this!
New features¶
- The latest version of the “map” function is ~3X faster than the one available in version 2.17 and 2.18
- The map function now supports the “-split” option, as well as “absmin” and “absmax” operations.
- In addition, it supports multiple chromosome sorting criterion by supplying a genome file that defines the expected chromosome order. Here is an example of how to run map with datasets having chromosomes sorted in “version” order, as opposed to the lexicographical chrom order that is the norm.
Version 2.18.2 (8-Jan-2014)¶
bedtools¶
The changes to bedtools reflect fixes to compilation errors, performance enhancements for smaller files, and a bug fix for BAM files that lack a formal header. Our current focus for the 2.19.* release is is on addressing some standing bug/enhancements and also in updating some of the other more widely used tools (e.g., coverage, map, and substract) to use the new API. We will also continue to look into ways to improve performance while hopefully reducing memory usage for algorithms that work with unsorted data (thanks to Ian Sudberry for the ping!).
pybedtools¶
Ryan Dale has updated pybedtools to accomodate bedtools 2.18.*, added unit tests, and provided new functionality and bug fixes. The details for this release are here: http://pythonhosted.org/pybedtools/changes.html
Version 2.18.1 (16-Dec-2013)¶
Fixes that address compilation errors with CLANG and force compilation of custom BamTools library.
Version 2.18.0 (13-Dec-2013)¶
The Google Code site is deprecated¶
It looks like the Google Code service is going the way of the venerable Google Reader. As such, we are moving the repository and all formal release tarballs to Github. We have started a new repository prosaically named “bedtools2”. The original bedtools repository will remain for historical purposes, but we created a new repository to distinguish the two code bases as they will become rather different over time.
[https://github.com/arq5x/bedtools2](https://github.com/arq5x/bedtools2)
We gutted the core API and algorithms¶
Much of Neil’s hard work has been devoted to completely rewriting the core file/stream writing API to be much more flexible in the adoption of new formats. In addition, he has substantially improved many of the core algorithms for detecting interval intersections.
Improved performance¶
The 2.18.0 release leverages these improvements in the “intersect” tool. Forthcoming releases will see the new API applied to other tools, but we started with intersect as it is the most widely used tool in the suite.
Performance with sorted datasets. The “chromsweep” algorithm we use for detecting intersections is now 60 times faster than when it was first release in version 2.16.2, and is 15 times than the 2.17 release. This makes the algorithm slightly faster that the algorithm used in the bedops bedmap tool. As an example, the following [figure](https://dl.dropboxusercontent.com/u/515640/bedtools-intersect-sorteddata.png) demonstrates the speed when intersecting GENCODE exons against 1, 10, and 100 million BAM alignments from an exome capture experiment. Whereas in version 2.16.2 this wuld have taken 80 minutes, it now takes 80 seconds.
Greater flexibility. In addition, BAM, BED, GFF/GTF, or VCF files are now automatically detected whether they are a file, stream, or FIFO in either compressed or uncompressed form. As such, one now longer has specify -abam when using BAM input as the “A” file with intersect. Moreover, any file type can be used for either the A or
the B file.
Better support for different chromosome sorting criteria¶
Genomic analysis is plagued by different chromosome naming and sorting conventions. Prior to this release,
the -sorted option in the intersect tool required that the chromosomes were sorted in alphanumeric
order (e.g. chr1, chr10, etc. or 1, 10, etc.). Starting with this release, we now simply require by default
that the records are GROUPED by chromosome and that within each chromosome group, the records are sorted by
chromosome position. This will allow greater flexibility.
One problem that can arise however, is if two different files are each grouped by chromosome, yet the two files follow a different chromosome order. In order to detect and enforce the same order, one can explicitly state the expected chromosome order through the use of a genome (aka chromsizes) file. Please see the documentation [here](http://bedtools.readthedocs.org/en/latest/content/tools/intersect.html#sorted-invoke-a-memory-efficient-algorithm-for-very-large-files) and [here](http://bedtools.readthedocs.org/en/latest/content/tools/intersect.html#g-define-an-alternate-chromosome-sort-order-via-a-genome-file) for examples.
New tools¶
1. The jaccard tool. While not exactly new, there have been improvements to the tool and there is finally
documentation. Read more here: http://bedtools.readthedocs.org/en/latest/content/tools/jaccard.html
- The
reldisttool. Details here: http://bedtools.readthedocs.org/en/latest/content/tools/reldist.html
3. The sample tool. Uses reservoir sampling to randomly sample a specified number of records from BAM, BED,
VCF, and GFF/GTF files.
Enhancements¶
- Improvements in the consistency of the output of the
mergetool. Thanks to @kcha.
2. A new -allowBeyondChromEnd option in the shuffle tool. Thanks to @stephenturner.
[docs](http://bedtools.readthedocs.org/en/latest/content/tools/shuffle.html#allowbeyondchromend-allow-records-to-extend-beyond-the-chrom-length)
- A new
-noOverlappingoption that prevents shuffled intervals from overlapping one another. Thanks to @brentp. [docs](http://bedtools.readthedocs.org/en/latest/content/tools/shuffle.html#nooverlapping-prevent-shuffled-intervals-from-overlapping) - Allow the user to specify the maximum number of shuffling attempts via the
-maxTriesoption in theshuffletool. - Various improvements to the documentation provided by manu different users. Thanks to all.
- Added the number of intersections (
n_intersections) to the Jaccard output. Thanks to @brentp. - Various improvements to the
tagtool. - Added the
-N(remove any) option to thesubtracttool.
Version 2.17.0 (3-Nov-2012)¶
New tools¶
We have added a new tool (bedtools “jaccard”) for measuring the Jaccard statistic between two interval files. The Jaccard stat measures the ratio of the length of the intersection over the length of the union of the two sets. In this case, the union is measured as the sum of the lengths of the intervals in each set minus the length of the intersecting intervals. As such, the Jaccard statistic provides a “distance” measure between 0 (no intersections) and 1 (self intersection). The higher the score, the more the two sets of intervals overlap one another. This tool was motivated by Favorov et al, 2012. For more details, see see PMID: 22693437.
We anticipate releasing other statistical measures in forthcoming releases.
New Features & enhancements¶
- The genome file drives the BAM header in “bedtools bedtobam”
- Substantially improvement the performance of the -sorted option in “bedtools intersect” and “bedtools map”. For many applications, bedtools is now nearly as fast as the BEDOPS suite when intersecting pre-sorted data. This improvement is thanks to Neil Kindlon, a staff scientist in the Quinlan lab.
- Tightened the logic for handling split (blocked) BAM and BED records
- Added ranged column selection to “bedtools groupby”. Thanks to Brent Pedersen”
- e.g., formerly “bedtools groupby -g 1,2,3,4,5”; now “-g 1-5”
- “bedtools getfasta” now properly extracts sequences based on blocked (BED12) records (e.g., exons from genes in BED12 format).
- “bedtools groupby” now allows a header line in the input.
- With -N, the user can now force the closest interval to have a different name field in “bedtools closest”
- With -A, the user can now force the subtraction of entire interval when any overlap exists in “bedtools subtract”.
- “bedtools shuffle” can now shuffle BEDPE records.
- Improved random number generation.
- Added -split, -s, -S, -f, -r options to “bedtools multicov”
- Improvements to the regression testing framework.
- Standardized the tag reporting logic in “bedtools bamtobed”
- Improved the auto-detection of VCF format. Thanks to Michael James Clark.
Bug fixes¶
- Fixed a bug in bedtobam’s -bed12 mode.
- Properly include unaligned BAM alignments with “bedtools intersect“‘s -v option.
- Fixed off by one error in “bedtools closest“‘s -d option
4.”bedtools bamtobed” fails properly for non-existent file.
- Corrected missing tab in “bedtools annotate“‘s header.
- Allow int or uint tags in “bedtools bamtobed”
- “bedtools flank” no longer attempts to take flanks prior to the start of a chromosome.
- Eliminated an extraneous tab from “bedtools window” -c.
- Fixed a corner case in the -sorted algorithm.
10.Prevent numeric overflow in “bedtools coverage -hist”
Version 2.14.1-3 (2-Nov-2011)¶
Bug Fixes¶
- Corrected the help for closestBed. It now correctly reads -io instead of -no.
- Fixed regression in closestBed injected in version 2.13.4 whereby B features to the right of an A feature were missed.
New tool¶
- Added the multiIntersectBed tool for reporting common intervals among multiple sorted BED/GFF/VCF files.
Version 2.13.4 (26-Oct-2011)¶
Bug Fixes¶
- The -sorted option (chromsweep) in intersectBed now obeys -s and -S. I had neglected to implement that. Thanks to Paul Ryvkin for pointing this out.
- The -split option was mistakenly splitting of D CIGAR ops.
- The Makefile was not including zlib properly for newer versions of GCC. Thanks to Istvan Albert for pointing this out and providing the solution.
Improvements¶
- Thanks to Jacob Biesinger for a new option (-D) in closestBed that will report _signed_ distances. Moreover, the new option allows fine control over whether the distances are reported based on the reference genome or based on the strand of the A or B feature. Many thanks to Jacob.
- Thanks to some nice analysis from Paul Ryvkin, I realized that the -sorted option was using way too much memory in certain cases where there is a chromosome change in a sorted BED file. This has been corrected.
Version 2.13.3 (30-Sept-2011)¶
Bug Fixes¶
- intersectBed detected, but did not report overlaps when using BAM input and -bed.
Other¶
- Warning that -sorted trusts, but does not enforce that data is actually sorted.
Version 2.13.2 (23-Sept-2011)¶
New algorithm¶
- Preliminary release of the chrom_sweep algorithm.
New options¶
- genomeCoverageBed no longer requires a genome file when working with BAM input. It instead uses the BAM header.
- tagBam now has a -score option for annotating alignments with the BED “scores” field in annotation files. This overrides the default behavior, which is to use the -labels associated with the annotation files passed in on the command line.
Bug fixes¶
- Correct a bug that prevented proper BAM support in intersectBed.
- Improved detection of GFF features with negative coordinates.
Version 2.13.1 (6-Sept-2011)¶
New options¶
- tagBam now has -s and -S options for only annotating alignments with features on the same and opposite strand, respectively.
- tagBam now has a -names option for annotating alignments with the “name” field in annotation files. This overrides the default behavior, which is to use the -labels associated with the annotation files passed in on the command line. Currently, this works well with BED files, but given the limited metadata support for GFF files, annotating with -names and GFF files may not work as well as wished, depending on the type of GFF file used.
Version 2.13.0 (1-Sept-2011)¶
New tools¶
1. tagBam. This tool annotates a BAM file with custom tag fields based on overlaps with BED/GFF/VCF files. For example:
$ tagBam -i aln.bam -files exons.bed introns.bed cpg.bed utrs.bed \
-tags exonic intonic cpg utr \
> aln.tagged.bam
For alignments that have overlaps, you should see new BAM tags like “YB:Z:exonic”, “YB:Z:cpg;utr” 2. multiBamCov. The new tool counts sequence coverage for multiple bams at specific loci defined in a BED/GFF/VCF file. For example:
$ multiBamCov -bams aln.1.bam aln.2.bam aln3.bam -bed exons.bed chr1 861306 861409 SAMD11 1 + 181 280 236 chr1 865533 865718 SAMD11 2 + 249 365 374 chr1 866393 866496 SAMD11 3 + 162 298 322
where the last 3 columns represent the number of alignments overlapping each interval from the three BAM file.
- The following options are available to control which types of alignments are are counted.
-q Minimum mapping quality allowed. Default is 0. -D Include duplicate-marked reads. Default is to count non-duplicates only -F Include failed-QC reads. Default is to count pass-QC reads only -p Only count proper pairs. Default is to count all alignments with MAPQ greater than the -q argument, regardless of the BAM FLAG field.
- nucBed. This new tool profiles the nucleotide content of intervals in a fasta file. The following information will be reported after each original BED/GFF/VCF entry:
- %AT content
- %GC content
- Number of As observed
- Number of Cs observed
- Number of Gs observed
- Number of Ts observed
- Number of Ns observed
- Number of other bases observed
- The length of the explored sequence/interval.
- The sequence extracted from the FASTA file. (optional, if -seq is used)
- The number of times a user defined pattern was observed. (optional, if -pattern is used.)
- For example:
- $ nucBed -fi ~/data/genomes/hg18/hg18.fa -bed simrep.bed | head -3 #1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_pct_at 8_pct_gc 9_num_A 10_num_C 11_num_G 12_num_T 13_num_N 14_num_oth 15_seq_len chr1 10000 10468 trf 789 + 0.540598 0.459402 155 96 119 98 0 0 468 chr1 10627 10800 trf 346 + 0.445087 0.554913 54 55 41 23 0 0 173
- One can also report the sequence itself:
- $ nucBed -fi ~/data/genomes/hg18/hg18.fa -bed simrep.bed -seq | head -3 #1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_pct_at 8_pct_gc 9_num_A 10_num_C 11_num_G 12_num_T 13_num_N 14_num_oth 15_seq_len 16_seq chr1 10000 10468 trf 789 + 0.540598 0.459402 155 96 119 98 0 0 468 ccagggg… chr1 10627 10800 trf 346 + 0.445087 0.554913 54 55 41 23 0 0 173 TCTTTCA…
- Or, one can count the number of times that a specific pattern occur in the intervals (reported as the last column):
- $ nucBed -fi ~/data/genomes/hg18/hg18.fa -bed simrep.bed -pattern CGTT | head #1_usercol 2_usercol 3_usercol 4_usercol 5_usercol 6_usercol 7_pct_at 8_pct_gc 9_num_A 10_num_C 11_num_G 12_num_T 13_num_N 14_num_oth 15_seq_len 16_user_patt_count chr1 10000 10468 trf 789 + 0.540598 0.459402 155 96 119 98 0 0 468 0 chr1 10627 10800 trf 346 + 0.445087 0.554913 54 55 41 23 0 0 173 0 chr1 10757 10997 trf 434 + 0.370833 0.629167 49 70 81 40 0 0 240 0 chr1 11225 11447 trf 273 + 0.463964 0.536036 44 86 33 59 0 0 222 0 chr1 11271 11448 trf 187 + 0.463277 0.536723 37 69 26 45 0 0 177 0 chr1 11283 11448 trf 199 + 0.466667 0.533333 37 64 24 40 0 0 165 0 chr1 19305 19443 trf 242 + 0.282609 0.717391 17 57 42 22 0 0 138 1 chr1 20828 20863 trf 70 + 0.428571 0.571429 10 7 13 5 0 0 35 0 chr1 30862 30959 trf 79 + 0.556701 0.443299 35 22 21 19 0 0 97 0
New options¶
Support for named pipes and FIFOs.
“-” is now allowable to indicate that data is being sent via stdin.
Multiple tools. Added new -S option to annotateBed, closestBed, coverageBed, intersectBed, pairToBed, subtractBed, and windowBed (-Sm). This new option does the opposite of the -s option: that is, overlaps are only processed if they are on _opposite_ strands. Thanks to Sol Katzman for the great suggestion. Very useful for certain RNA-seq analyses.
coverageBed. Added a new -counts option to coverageBed that only reports the count of overlaps, instead of also computing fractions, etc. This is much faster and uses much less memory.
fastaFromBed. Added a new -full option that uses the full BED entry when naming each output sequence. Also removed the -fo option such that all output is now written to stdout.
- genomeCoverageBed.
- Added new -scale option that allows the coverage values to be scaled by a constant. Useful for normalizing coverage with RPM, RPKM, etc. Thanks to Ryan Dale for the useful suggestion.
- Added new -5, -3, -trackline, -trackopts, and -dz options. Many thanks to Assaf Gordon for these improvements.
- -5: Calculate coverage of 5” positions (instead of entire interval) -3: Calculate coverage of 3” positions (instead of entire interval). -trackline: Adds a UCSC/Genome-Browser track line definition in the first line of the output. -trackopts: rites additional track line definition parameters in the first line. -dz: Report the depth at each genome position with zero-based coordinates, instead of zero-based.
- closestBed. See below, thanks to Brent Pedersen, Assaf Gordon, Ryan Layer and Dan Webster for the helpful discussions.
- closestBed now reports _all_ features in B that overlap A by default. This allows folks to decide which is the “best” overlapping feature on their own. closestBed now has a “-io” option that ignores overlapping features. In other words, it will only report the closest, non-overlapping feature.
An example:
$ cat a.bed chr1 10 20
$ cat b.bed chr1 15 16 chr1 16 40 chr1 100 1000 chr1 200 1000
$ bin/closestBed -a a.bed -b b.bed chr1 10 20 chr1 15 16 chr1 10 20 chr1 16 40
$ bin/closestBed -a a.bed -b b.bed -io chr1 10 20 chr1 100 1000
Updates¶
- Updated to the latest version of BamTools. This allows greater functionality and will facilitate new options and tools in the future.
Bug Fixes¶
- GFF files cannot have zero-length features.
- Corrected an erroneous check on the start coordinates in VCF files. Thanks to Jan Vogel for the correction.
- mergeBed now always reports output in BED format.
- Updated the text file Tokenizer function to yield 15% speed improvement.
- Various tweaks and improvements.
Version 2.12.0 (April-3-2011)¶
New Tool¶
- Added new tool called “flankBed”, which allows one to extract solely the flanking regions that are upstream and downstream of a given feature. Unlike slopBed, flankBed does not include the original feature itself. A new feature is created for each flabking region. For example, imagine the following feature:
chr1 100 200
The following would create features for solely the 10 bp regions flanking this feature. $ bin/flankBed -i a.bed -b 10 -g genomes/human.hg18.genome chr1 90 100 chr1 200 210
In contrast, slopBed would return: bin/slopBed -i a.bed -b 10 -g genomes/human.hg18.genome chr1 90 210
FlankBed has all of the same features as slopBed.
New Features¶
1. Added new “-scores” feature to mergeBed. This allows one to take the sum, min, max, mean, median, mode, or antimode of merged feature scores. In addition, one can use the “collapse” operation to get a comma-separated list of the merged scores. 2. mergeBed now tolerates multiple features in a merged block to have the same feature name. 3. Thanks to Erik Garrison’s “fastahack” library, fastaFromBed now reports its output in the order of the input file. 4. Added a “-n” option to bed12ToBed6, which forces the score field to be the 1-based block number from the original BED12 feature. This is useful for tracking exon numbers, for example. 5. Thanks to Can Alkan, added a new “-mc” option to maskFastaFromBed that allows one to define a custom mask character, such as “X” (-n X).
Bug Fixes¶
- Thanks to Davide Cittaro, intersectBed and windowBed now properly capture unmapped BAM alignments when using the “-v” option.
- ClosestBed now properly handles cases where b.end == a.start
- Thanks to John Marshall, the default constructors are much safer and less buggy.
- Fixed bug in shuffleBed that complained about a lack of -incl and -excl.
- Fixed bug in shuffleBed for features that would go beyond the end of a chromosome.
- Tweaked bedToIgv to make it more Windows friendly.
Version 2.11.2 (January-31-2010)¶
Fixed a coordinate reporting bug in coverageBed. Added “max distance (-d)” argument back to the new implementation of mergeBed.
Version 2.11.0 (January-21-2010)¶
Enhancements:¶
- Support for zero length features (i.e., start = end) - For example, this allows overlaps to be detected with insertions in the reference genome, as reported by dbSNP.
- Both 8 and 9 column GFF files are now supported.
- slopBed can now extend the size of features by a percentage of it’s size (-pct) instead of just a fixed number of bases.
- Two improvements to shuffleBed: 3a. A -f (overlapFraction) parameter that defines the maximum overlap that a randomized feature can have with an -excl feature. That is, if a chosen locus has more than -f overlap with an -excl feature, a new locus is sought. 3b. A new -incl option (thanks to Michael Hoffman and Davide Cittaro) that, defines intervals in which the randomized features should be placed. This is used instead of placing the features randomly in the genome. Note that a genome file is still required so that a randomized feature does not go beyond the end of a chromosome.
- bamToBed can now optionally report the CIGAR string as an additional field.
- pairToPair can now report the entire paired feature from the B file when overlaps are found.
- complementBed now reports all chromosomes, not just those with features in the BED file.
- Improved randomization seeding in shuffleBed. This prevents identical output for runs of shuffleBed that occur in the same second (often the case).
Bug Fixes:¶
- Fixed the “BamAlignmentSupportData is private” compilation issue.
- Fixed a bug in windowBed that caused positions to run off the end of a chromosome.
Major Changes:¶
- The groupBy command is now part of the filo package (https://github.com/arq5x/filo) and will no longer be distributed with BEDTools.
Version 2.10.0 (September-21-2010)¶
New tools¶
1. annotateBed. Annotates one BED/VCF/GFF file with the coverage and number of overlaps observed from multiple other BED/VCF/GFF files. In this way, it allows one to ask to what degree one feature coincides with multiple other feature types with a single command. For example, the following will annotate the fraction of the variants in variants.bed that are covered by genes, conservaed regions and know variation, respectively. $ annotateBed -i variants.bed -files genes.bed conserv.bed known_var.bed
This tool was suggested by Can Alkan and was motivated by the example source code that he kindly provided.
New features¶
- New frequency operations (freqasc and freqdesc) added to groupBy. These operations report a histogram of the frequency that each value is observed in a given column.
- Support for writing uncompressed bam with the -ubam option.
- Shorthand arguments for groupBy (-g eq. -grp, -c eq. -opCols, -o eq. -opCols).
- In addition, all BEDTools that require only one main input file (the -i file) will assume that input is coming from standard input if the -i parameter is ignored.
Bug fixes¶
- Increased the precision of the output from groupBy.
Version 2.9.0 (August-16-2010)¶
New tools¶
- unionBedGraphs. This is a very powerful new tool contributed by Assaf Gordon from CSHL. It will combine/merge multiple BEDGRAPH files into a single file, thus allowing comparisons of coverage (or any text-value) across multiple samples.
New features¶
- New “distance feature” (-d) added to closestBed by Erik Arner. In addition to finding the closest feature to each feature in A, the -d option will report the distance to the closest feature in B. Overlapping features have a distance of 0.
- New “per base depth feature” (-d) added to coverageBed. This reports the per base coverage (1-based) of each feature in file B based on the coverage of features found in file A. For example, this could report the per-base depth of sequencing reads (-a) across each capture target (-b).
Bug Fixes¶
- Fixed bug in closestBed preventing closest features from being found for A features with start coordinates < 2048000. Thanks to Erik Arner for pointing this out.
- Fixed minor reporting annoyances in closestBed. Thanks to Erik Arner.
- Fixed typo/bug in genomeCoverageBed that reported negative coverage owing to numeric overflow. Thanks to Alexander Dobin for the detailed bug report.
- Fixed other minor parsing and reporting bugs/annoyances.
Version 2.8.3 (July-25-2010)¶
- Fixed bug that caused some GFF files to be misinterpreted as VCF. This prevented the detection of overlaps.
- Added a new “-tag” option in bamToBed that allows one to choose the _numeric_ tag that will be used to populate the score field. For example, one could populate the score field with the alignment score with “-tag AS”.
- Updated the BamTools API.
Version 2.8.2 (July-18-2010)¶
- Fixed a bug in bedFile.h preventing GFF strands from being read properly.
- Fixed a bug in intersectBed that occasionally caused spurious overlaps between BAM alignments and BED features.
- Fixed bug in intersectBed causing -r to not report the same result when files are swapped.
- Added checks to groupBy to prevent the selection of improper opCols and groups.
- Fixed various compilation issues, esp. for groupBy, bedToBam, and bedToIgv.
- Updated the usage statements to reflect bed/gff/vcf support.
- Added new fileType functions for auto-detecting gzipped or regular files. Thanks to Assaf Gordon.
Version 2.8.1 (July-05-2010)¶
- Added bedToIgv.
Version 2.8.0 (July-04-2010)¶
- Proper support for “split” BAM alignments and “blocked” BED (aka BED12) features. By using the “-split” option, intersectBed, coverageBed, genomeCoverageBed, and bamToBed will now correctly compute overlaps/coverage solely for the “split” portions of BAM alignments or the “blocks” of BED12 features such as genes.
- Added native support for the 1000 Genome Variant Calling Format (VCF) version 4.0.
- New bed12ToBed6 tool. This tool will convert each block of a BED12 feature into discrete BED6 features.
- Useful new groupBy tool. This is a very useful new tool that mimics the “groupBy” clause in SQL. Given a file or stream that is sorted by the appropriate “grouping columns”, groupBy will compute summary statistics on another column in the file or stream. This will work with output from all BEDTools as well as any other tab-delimited file or stream. Example summary operations include: sum, mean, stdev, min, max, etc. Please see the help for the tools for examples. The functionality in groupBy was motivated by helpful discussions with Erik Arner at Riken.
- Improvements to genomeCoverageBed. Applied several code improvements provided by Gordon Assaf at CSHL. Most notably, beyond the several efficiency and organizational changes he made, he include a “-strand” option which allows one to specify that coverage should only be computed on either the “+” or the “-” strand.
- Fixed a bug in closestBed found by Erik Arner (Riken) which incorrectly reported “null” overlaps for features that did not have a closest feature in the B file.
- Fixed a careless bug in slopBed also found by Erik Arner (Riken) that caused an infinite loop when the “-excl” option was used.
- Reduced memory consumption by ca. 15% and run time by ca. 10% for most tools.
- Several code-cleanliness updates such as templated functions and common tyedefs.
- Tweaked the genome binning approach such that 16kb bins are the most granular.
Version 2.7.1 (May-06-2010)¶
Fixed a typo that caused some compilers to fail on closestBed.
Version 2.7.0 (May-05-2010)¶
General: 1. “Gzipped” BED and GFF files are now supported as input by all BEDTools. Such files must end in “.gz”. 2. Tools that process BAM alignments now uniformly compute an ungapped alignment end position based on the BAM CIGAR string. Specifically, “M”, “D” and “N” operations are observed when computing the end position. 3. bamToBed requires the BAM file to be sorted/grouped by read id when creating BEDPE output. This allows the alignments end coordinate for each end of the pair to be properly computed based on its CIGAR string. The same requirement applies to pairToBed. 4. Updated manual. 5. Many silent modifications to the code that improve clarity and sanity-checking and facilitate future additions/modifications.
New Tools: 1. bedToBam. This utility will convert BED files to BAM format. Both “blocked” (aka BED12) and “unblocked” (e.g. BED6) formats are acceptable. This allows one to, for example, compress large BED files such as dbSNP into BAM format for efficient visualization.
- Changes to existing tools:
- intersectBed
- Added -wao option to report 0 overlap for features in A that do not intersect any features in B. This is an extension of the -wo option.
- bamToBed
- Requires that BAM input be sorted/grouped by read name.
- pairToBed
- Requires that BAM input be sorted/grouped by read name.
- Allows use of minimum mapping quality or total edit distance for score field.
- windowBed
- Now supports BAM input.
- genomeCoverageBed
- -bga option. Thanks to Gordon Assaf for the suggestion.
- Eliminated potential seg fault.
- Acknowledgements:
- Gordon Assaf: for suggesting the -bga option in genomeCoverageBed and for testing the new bedToBam utility.
- Ivan Gregoretti: for helping to expedite the inclusion of gzip support.
- Can Alkan: for suggesting the addition of the -wao option to intersectBed.
- James Ward: for pointing out that bedToBam did not need to create “dummy” seq and qual entries.
Version 2.6.1 (Mar-29-2010)¶
- Fixed a careless command line parsing bug in coverageBed.
Version 2.6.0 (Mar-23-2010)¶
Specific improvements / additions to tools¶
- intersectBed. Added an option (-wo) that reports the number of overlapping bases for each intersection b/w A and B files. Not sure why this wasn’t added sooner; it’s obvious.
2. coverageBed - native BAM support - can now report a histogram (-hist) of coverage for each feature in B. Useful for exome sequencing projects, for example. Thanks for the excellent suggestion from Jose Bras - faster
3. genomeCoverageBed - native BAM support - can now report coverage in BEDGRAPH format (-bg). Thanks for the code and great suggestion from Gordon Assaf, CSHL.
4. bamToBed - support for “blocked” BED (aka BED12) format. This facilitates the creation of BED entries for “split” alignments (e.g. RNAseq or SV). Thanks to Ann Loraine, UNCC for test data to support this addition.
5. fastaFromBed - added the ability to extract sequences from a FASTA file according to the strand in the BED file. That is, when “-” the extracted sequence is reverse complemented. Thanks to Thomas Doktor, U. of Southern Denmark for the code and suggestion.
6. *NEW* overlap - newly added tool for computing the overlap/distance between features on the same line.For example:
$ cat test.out chr1 10 20 A chr1 15 25 B chr1 10 20 C chr1 25 35 D $ cat test.out | overlaps -i stdin -cols 2,3,6,7 chr1 10 20 A chr1 15 25 B 5 chr1 10 20 C chr1 25 35 D -5
Bug fixes¶
- Fixed a bug in pairToBed when comparing paired-end BAM alignments to BED annotations and using the “notboth” option.
- Fixed an idiotic bug in intersectBed that occasionally caused segfaults when blank lines existed in BED files.
- Fixed a minor bug in mergeBed when using the -nms option.
General changes¶
- Added a proper class for genomeFiles. The code is much cleaner and the tools are less sensitive to minor problems with the formatting of genome files. Per Gordon Assaf’s wise suggestion, the tools now support “chromInfo” files directly downloaded from UCSC. Thanks Gordon—I disagreed at first, but you were right.
- Cleaned up some of the code and made the API a bit more streamlined. Will facilitate future tool development, etc.
Version 2.5.4 (Mar-3-2010)¶
- Fixed an insidious bug that caused malform BAM output from intersectBed and pairToBed. The previous BAM files worked fine with samtools as BAM input, but when piped in as SAM, there was an extra tab that thwarted conversion from SAM back to BAM. Many thanks to Ivan Gregoretti for reporting this bug. I had never used the BAM output in this way and thus never caught the bug!
Version 2.5.3 (Feb-19-2010)¶
- Fixed bug to “re-allow” track and “browser” lines.
- Fixed bug in reporting BEDPE overlaps.
- Fixed bug when using type “notboth” with BAM files in pairToBed.
- When comparing BAM files to BED/GFF annotations with intersectBed or pairToBed, the __aligned__ sequence is used, rather than the __original__ sequence.
- Greatly increased the speed of pairToBed when using BAM alignments.
- Fixed a bug in bamToBed when reporting edit distance from certain aligners.
Version 2.5.2 (Feb-2-2010)¶
- The start and end coordinates for BED and BEDPE entries created by bamToBed are now based on the __aligned__ sequence, rather than the original sequence. It’s obvious, but I missed it originally…sorry.
- Added an error message to mergeBed preventing one from using “-n” and “-nms” together.
- Fixed a bug in pairToBed that caused neither -type “notispan” nor “notospan” to behave as described.
Version 2.5.1 (Jan-28-2010)¶
- Fixed a bug in the new GFF/BED determinator that caused a segfault when start = 0.
Version 2.5.0 (Jan-27-2010)¶
- Added support for custom BED fields after the 6th column.
- Fixed a command line parsing bug in pairToBed.
- Improved sanity checking.
Version 2.4.2 (Jan-23-2010)¶
- Fixed a minor bug in mergeBed when -nms and -s were used together.
- Improved the command line parsing to prevent the occasional segfault.
Version 2.4.1 (Jan-12-2010)¶
- Updated BamTools libraries to remove some compilation issues on some systems/compilers.
Version 2.4.0 (Jan-11-2010)¶
- Added BAM support to intersectBed and pairToBed
- New bamToBed feature.
- Added support for GFF features
- Added support for “blocked” BED format (BED12)
- Wrote complete manual and included it in distribution.
- Fixed several minor bugs.
- Cleaned up code and improved documentation.
Version 2.3.3 (12/17/2009)¶
Rewrote complementBed to use a slower but much simpler approach. This resolves several bugs with the previous logic.
Version 2.3.2 (11/25/2009)¶
- Fixed a bug in subtractBed that prevent a file from subtracting itself when the following is used:
- $ subtractBed -a test.bed -b test.bed
Version 2.3.1 (11/19/2009)¶
Fixed a typo in closestBed that caused all nearby features to be returned instead of just the closest one.
Version 2.3.0 (11/18/2009)¶
- Added four new tools:
- shuffleBed. Randomly permutes the locations of a BED file among a genome. Useful for testing for significant overlap enrichments.
- slopBed. Adds a requested number of base pairs to each end of a BED feature. Constrained by the size of each chromosome.
- maskFastaFromBed. Masks a FASTA file based on BED coordinates. Useful making custom genome files from targeted capture experiment, etc.
- pairToPair. Returns overlaps between two paired-end BED files. This is great for finding structural variants that are private or shared among samples.
- Increased the speed of intersectBed by nearly 50%.
- Improved corrected some of the help messages.
- Improved sanity checking for BED entries.
Version 2.2.4 (10/27/2009)¶
1. Updated the mergeBed documentation to describe the -names option which allows one to report the names of the features that were merged (separated by semicolons).
Version 2.2.3 (10/23/2009)¶
1. Changed windowBed to optionally define “left” and “right” windows based on strand. For example by default, -l 100 and -r 500 will add 100 bases to the left (lower coordinates) of a feature in A when scanning for hits in B and 500 bases to the right (higher coordinates).
However if one chooses the -sw option (windows bases on strandedness), the behavior changes. Assume the above example except that a feature in A is on the negative strand (“-“). In this case, -l 100, -r 500 and -sw will add 100 bases to the right (higher coordinates) and 500 bases to the left (lower coordinates).
In addition, there is a separate option (-sm) that can optionally force hits in B to only be tracked if they are on the same strand as A.
*NOTE: This replaces the previous -s option and may affect existing pipelines*.
Version 2.2.2 (10/20/2009)¶
- Improved the speed of genomeCoverageBed by roughly 100 fold. The memory usage is now less than 2.0 Gb.
Version 2.2.1¶
1. Fixed a very obvious bug in subtractBed that caused improper behavior when a feature in A was overlapped by more than one feature in B. Many thanks to folks in the Hannon lab at CSHL for pointing this out.
Version 2.2.0¶
Notable changes in this release¶
- coverageBed will optionally only count features in BED file A (e.g. sequencing reads) that overlap with
the intervals/windows in BED file B on the same strand. This has been requested several times recently and facilitates CHiP-Seq and RNA-Seq experiments.
- intersectBed can now require a minimum __reciprocal__ overlap between intervals in BED A and BED B. For example,
previously, if one used -f 0.90, it required that a feature in B overlap 90% of the feature in A for the “hit” to be reported. If one adds the -r (reciprocal) option, the hit must also cover 90% of the feature in B. This helps to exclude overlaps between say small features in A and large features in B:
A ========== B ******************************************************
-f 0.50 (Reported), whereas -f 0.50 -r (Not reported)
- The score field has been changed to be a string. While this deviates from the UCSC definition, it allows one to track
much more meaningful information about a feature/interval. For example, score could now be:
7.31E-05 (a p-value) 0.334577 (mean enrichment) 2:2.2:40:2 (several values encoded in a string)
- closestBed now, by default, reports __all__ intervals in B that overlap equally with an interval in A. Previously, it
merely reported the first such feature that appeared in B. Here’s a cartoon explaining the difference.
Several other minor changes to the algorithms have been made to increase speed a bit.
Version 2.1.2¶
- Fixed yet another bug in the parsing of “track” or “browser” lines. Sigh…
- Change the “score” column (i.e. column 5) to b stored as a string. While this deviates from the UCSC convention, it allows significantly more information to be packed into the column.
Version 2.1.1¶
- Added limits.h to bedFile.h to fix compilation issues on some systems.
- Fixed bug in testing for “track” or “browser” lines.
Version 2.1.0¶
- Fixed a bug in peIntersectBed that prevented -a from being correctly handled when passed via stdin.
- Added new functionality to coverageBed that calculates the density of coverage.
- Fixed bug in geneomCoverageBed.
Version 2.0.1¶
- Added the ability to retain UCSC browser track/browser headers in BED files.
Version 2.0¶
- Sped up the file parsing. ~10-20% increase in speed.
- Created reportBed() as a common method in the bedFile class. Cleans up the code quite nicely.
- Added the ability to compare BED files accounting for strandedness.
- Paired-end intersect.
- Fixed bug that prevented overlaps from being reported when the overlap fraction requested is 1.0
Version 1.2, 04/27/2009.¶
- Added subtractBed.
- Fixed bug that prevented “split” overlaps from being reported.
- Prevented A from being reported if >=1 feature in B completely spans it.
- Added linksBed.
- Added the ability to define separate windows for upstream and downstream to windowBed.
Version 1.1, 04/23/2009.¶
Initial release.

{kind=link}
{kind=link}